KAS-Analyzer: a comprehensive framework for exploring KAS-seq data
Author: Ruitu Lyu, Chemistry department, the University of Chicago
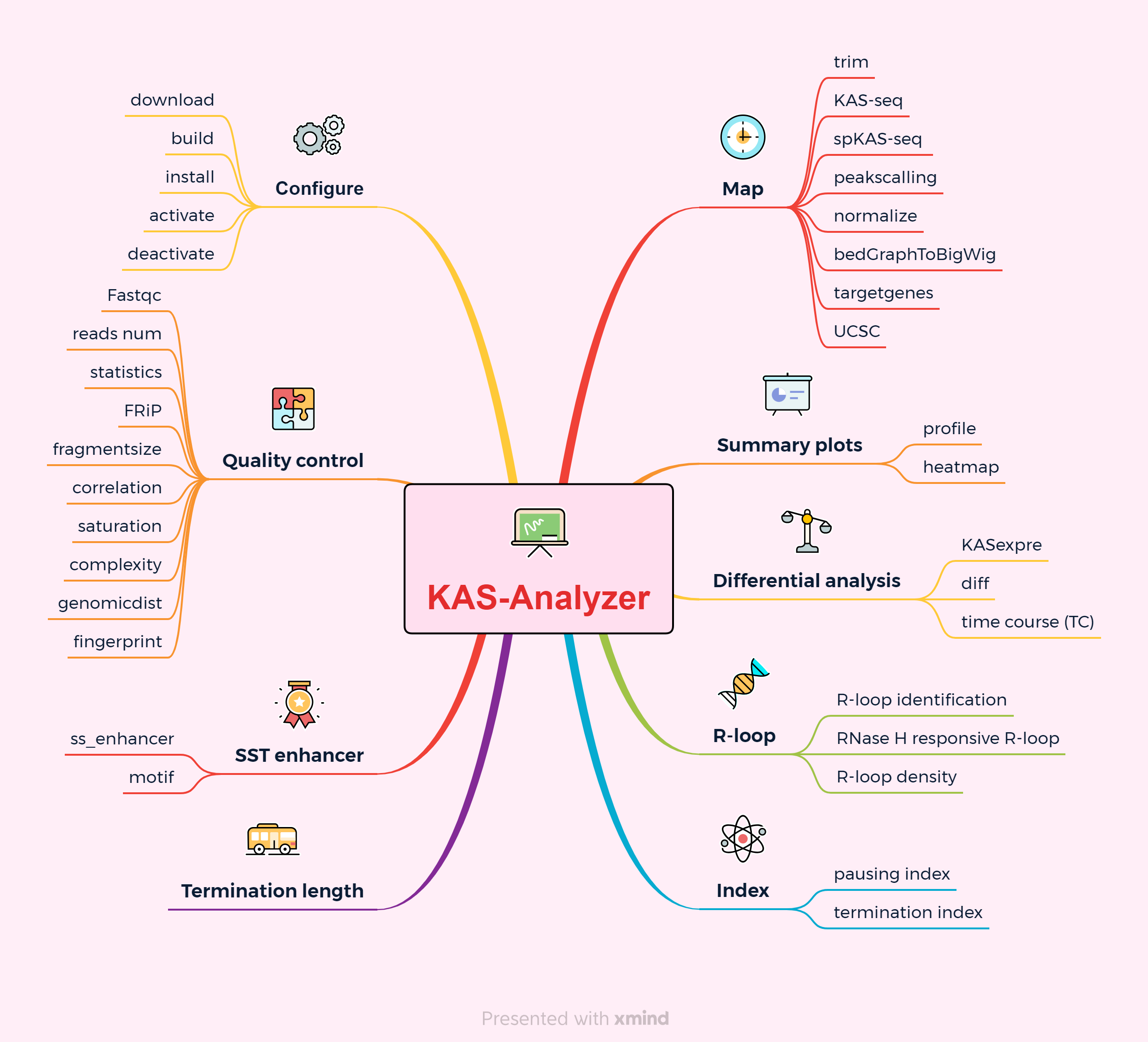
Introduction
Here we introduce KAS-Analyzer, a newly designed, comprehensive methodological framework aimed at simplifying the analysis and interpretation of KAS-seq data. In addition to standard analyses, KAS-Analyzer offers many novel features specifically tailored for KAS-seq data, including, but not limited to: calculation of termination length and index, identification of single-stranded transcribing (SST) enhancers, high-resolution mapping of R-loops, and time-course differential RNA polymerase activity analysis.
KAS-Analyzer is still on active development and the source code is hosted on GitHub.
Installation
Install by cloning KAS-Analyzer git repository on github:
You can install KAS-Analyzer on command line (linux/mac) by cloning git repository on github:
$ git clone https://github.com/Ruitulyu/KAS-Analyzer.git
$ cd KAS-Analyzer
$ bash ./setup.sh
# If anaconda or miniconda was not installed on your system. #OPTIONAL
$ KAS-Analyzer install -conda
# Make sure you have Mamba installed. If not, you can install it in your base Conda environment with the following command.
$ conda install mamba -c conda-forge
# Install 'KAS-Analyzer' environment using mamba.
$ mamba env create -f ./environment.yml
# or
$ KAS-Analyzer install -KAS-Analyzer
# Activate conda 'KAS-Analyzer' environment.
$ conda activate KAS-Analyzer
Overview
KAS-Analyzer is a collection of command line tools for KAS-seq or strand specific KAS-seq(spKAS-seq) data analysis.
Version: v2.0
About: KAS-Analyzer is mainly developed by Ruitu Lyu, postdoc fellow in Prof. Chuan He's group at the University of Chicago.
Docs: https://ruitulyu.github.io/KAS-Analyzer/
Code: https://github.com/Ruitulyu/KAS-Analyzer
Mail: https://github.com/Ruitulyu/KAS-Analyzer/discussions
Usage:
KAS-Analyzer sub-command [options]
The KAS-Analyzer sub-commands include:
Configure
download Downlaod the index of reference genome for aligners (bowtie2, bwa).
build Build the index of reference genome for aligners (bowtie2, bwa).
install Install and check the 'KAS-Analyzer' conda environment; check the installation of tools.
uninstall Uninstall the 'KAS-Analyzer' conda environment.
activate Activate the 'KAS-Analyzer' conda environment.
deactivate Deactivate the 'KAS-Analyzer' conda environment.
Fastqc
fastqc Generate basic quality control metrics for KAS-seq data.
readsnum Calcuate the reads number of raw fastq files.
statistics Calculate the mapping statistics for KAS-seq data.
FRiP Calculate the fraction of reads in peaks.
fragmentsize Measure the fragment size of paired-end KAS-seq data.
correlation Calculate the correlation coefficient and pvalue, generate correlation plot for replicates of KAS-seq data.
saturation Perform saturation analysis for KAS-seq data.
complexity Calculate the complexity metric for (sp)KAS-seq data, including the PCR Bottlenecking Coefficient and Non-Redundant Fraction (NRF).
genomicdist Visualize the genomic distribution for KAS-seq peaks (table and plot).
fingerprint Plot fingerprint for KAS-seq data.
Mapping
trim Trim adapter and low quality sequence, perform quality control for raw KAS-seq data.
KAS-seq Align KAS-seq data to the reference genome, deduplicate mapped reads, and generate several files with maped reads (bam, bed and bedGraph).
spKAS-seq Align strand specific KAS-seq (spKAS-seq) data. Note: we strongly recommend paired-end sequencing for spKAS-seq data to accurately measure the fragment size.
peakscalling Call broad or sharp peaks for KAS-seq data.
normalize Normalize KAS-seq data with bedGraph density files.
ToBigWig Convert normalized bedGraph files to bigWig files.
targetgenes Define target or associated genes (promoter, genebody, terminator or gene) of KAS-seq peaks, R-loops or enhancers loci."
UCSC Generate bedGraph files ready for submitting to UCSC genome browser.
Summary plots
profile Generate metagene profile for KAS-seq data (normalized bigWig files are needed).
heatmap Generate heatmap for KAS-seq data (normalized bigWig files are needed).
Differential KAS-seq analysis
diff Perform differential KAS-seq analysis on promoter, genebody, gene, bin or custom regions.
TC Perform 'case-only' or 'case-control' differential time course(TC) analysis for (sp)KAS-seq data.
PCA Perform and plot PCA analysis for (sp)KAS-seq data.
R-loops
R-loop Identify R-loops regions with spKAS-seq data.
RNaseH Identify R-loops regions sensitive to RNase H treatment.
Single-stranded enhancers identification
SST_enhancer Identify the single-stranded transcribing enhancers (SST_enhancers).
motif Identify enriched TF binding motifs on single-stranded transcribing enhancers.
Termination length
termilength Calculate the transcription termination length of protein coding genes.
KAS-seq index
index Calculate the pausing or termination index.
KASindex Calculate KAS-seq index on promoter, genebody, genes or custom regions.
General help
--help Print this help menu.
--version Print the version of KAS-Analyzer you are using.
--contact Feature requests, bugs, mailing lists, etc.
Tutorial
Configure sub-commands:
download
Downlaod the index of reference genome for aligners (bowtie2 or bwa).
usage: KAS-Analyzer download [ -l ] [ -h ] [ -a aligner ] [ -g assembly id ] [ -d directory to save index of aligner ]
Example: KAS-Analyzer download -a bowtie2 -g hg19 -d /Software/reference_genome/
-l list all of the available aligner index for reference genomes.
-a [aligner]: aligner name you want to use, e.g. bowtie, bowtie2 or bwa. REQUIRED.
-g [assembly id]: reference genome assembly id, e.g. Human: hg18, hg19, hg38; Mouse: mm9,mm10,mm39; Fruitfly: dm3, dm6; Rat: rn6, rn7; C.elegans: ce10, ce11; Zebra fish: danRer10, danRer11. REQUIRED.
-d [directory to save index of aligner]: directory to save downloaded reference genome index. REQUIRED.
-h\-help: print this help and exit.
build
Build the index of reference genome for aligners (bowtie2 or bwa).
usage: KAS-Analyzer build [ -h ] [ -a aligner ] [ -g genome fasta ] [ -p index prefix ] [ -t threads ] [ -d index dir ]
Example: KAS-Analyzer build -a bowtie2 -g ./genome.fa -p hg19 -t 10 -d /Software/hg19_Bowtie2Index/
-a [aligner]: please specify aligner name you want to use, e.g. bowtie, bowtie2 or bwa. REQUIRED.
-g [genome fasta]: please input the path of reference genome fasta file. REQUIRED.
-t [threads]: please specify the number of threads. DEFAULT: 1.
-p [index prefix]: please input the prefix (basename) of the aligners' (bowtie, bowtie2 or bwa) reference genome index. DEFAULT: basename of fasta file .
-d [index dir]: directory to save newly built reference genome index. REQUIRED.
-h\-help: print this help and exit.
install
Install and check the ‘KAS-Analyzer’ conda environment; check the installation of tools.
usage: KAS-Analyzer install [ -h\--help ] [ -conda ] [ -check ] [ -t tools ] [ -KAS-Analyzer]
Example: KAS-Analyzer install or KAS-Analyzer install -check
-conda: check the installation or install anaconda in your computer.
-check: list the installed and uninstalled tools.
-KAS-Analyzer: install and configure the KAS-Analyzer conda environment.
-t [tools]: check the installation of specific tool, if not, will install automaticially.
-h\-help: print this help and exit.
Note: this subcommand is used to install conda environment and specific tool that needed in KAS-Analyzer.
uninstall
Uninstall ‘KAS-Analyzer’ conda environment.
usage: KAS-Analyzer uninstall
activate
Activate ‘KAS-Analyzer’ conda environment.
usage: conda activate KAS-Analyzer
deactivate
Deactivate ‘KAS-Analyzer’ conda environment.
usage: conda deactivate
Fastqc sub-commands:
fastqc
Generate basic quality control metrics for KAS-seq data.
usage: KAS-Analyzer fastqc [ -h/--help ] [ -t threads ] [ -c contaminants ] [ -o output dir ] [ -k KAS-seq ]
Example: nohup KAS-Analyzer fastqc -t 10 -k KAS-seq.rep1.fastq.gz,KAS-seq.rep2.fastq.gz,KAS-seq.rep3.fastq.gz &
-t [threads]: please input number of threads to be used for quality control check. DEFAULT: 1.
-c [contaminants]: please specify a file which contains the list of contaminants (format: name[tab]sequence) to screen overrepresented sequences against. DEFAULT: no.
-o [output dir]: please specify the output directory with output files.
-k [KAS-seq]: please input the KAS-seq data that you want to know the quality control, like sequencing quality, duplicates, contaminants (adapter sequence).
-h/--help: print this help and exit.
Note: The 'KAS-Analyzer fastqc' shell script is applied to check quality control and identify a potential type of problem in your KAS-seq data in non-interactive mode. It mainly invoke FASTQC, please refer to the FASTQC official website for more information.
readsnum
Calculate the reads number of raw sequencing files.
usage: KAS-Analyzer readsnum [ -h/--help ] [ -o prefix ] [ -f format ]
Example: nohup KAS-Analyzer readsnum -o KAS-seq_reads_num -f fastq.gz &
-o [prefix]: please specify the prefix (basename) of 'KAS-Analyzer readsnum' output files. REQUIRED.
-f [format]: please specify the format of raw reads data. e.g. fastq, fq, fastq.gz, fasta, fa or fa.gz. REQUIRED.
-h/--help: print this help and exit.
Note: The 'KAS-Analyzer readsnum' shell script is applied to calculate the reads number of raw sequencing files.
statistics
Generate the table containing (sp)KAS-seq mapping statistics.
usage: KAS-Analyzer statistics [ -h/--help ] [ -o prefix ] [ -l labels ] [ -s summary folder ]
Example: nohup KAS-Analyzer statistics -o KAS-seq_statistics -l labels.txt -s /absolute path/Summary/ &
-o [prefix]: please input the prefix (basename) of 'KAS-Analyzer statistics' output files. REQUIRED.
-l [labels]: please input the txt file containing labels of (sp)KAS-seq summary file generated by 'KAS-Analyzer (sp)KAS-seq'. DEFAULT: basename of summary file.
KAS-seq.rep1
KAS-seq.rep2
KAS-seq.rep3
KAS-seq.rep4 ---labels.txt
-s [summary folder or summary.txt]: please input the absolute path of folder with summary files or the txt file containing the summary files. e.g. -s summary.txt or /absolute path of summary folder. REQUIRED.
WT.rep1.KAS-seq_mapping_summary.txt or /absolute path/Summary/
WT.rep2.KAS-seq_mapping_summary.txt
KO.rep1.KAS-seq_mapping_summary.txt
KO.rep2.KAS-seq_mapping_summary.txt ---summary.txt
-h/--help: print this help and exit.
Note: The 'KAS-Analyzer statistics' shell script is applied to generate the table containing (sp)KAS-seq mapping statistics.
FRiP
Calculate and plot fraction of reads in peaks (FRiP) scores.
Usage: KAS-Analyzer FRiP [ -h/--help ] [ -o prefix ] [ -p peaks ] [ -l labels ] [ -k KAS-seq ]
Example: nohup KAS-Analyzer FRiP -o KAS-seq_FRiP -p peaks_files.txt -l labels.txt -k KAS-seq.txt &
-o [prefix]: please input the prefix (basename) of 'KAS-Analyzer FRiP' output files. REQUIRED.
-p [peaks]: please input the text file containing the peaks files. REQUIRED.
Example:
KAS-seq_WT_rep1_peaks.bed
KAS-seq_WT_rep2_peaks.bed
KAS-seq_KO_rep1_peaks.bed
KAS-seq_KO_rep2_peaks.bed ---peaks_files.txt
-l [labels]: please input the text file containing the labels of KAS-seq or spKAS-seq data that show in fraction of reads in peaks (FRiP) plot. DEFAULT: basename of KAS-seq files.
Example:
WT_rep1
WT.rep2
KO.rep1
KO.rep2 ---labels.txt
-k [KAS-seq]: please input the text file containing bed files (uniquely mapped reads used for 'KAS-Analyzer peakcalling'), which are used to calcuate fraction of reads in peaks (FRiP) score. The order and number of (sp)KAS-seq data should be the consistent with the labels file. REQUIRED.
Example:
KAS-seq_WT_rep1.bed
KAS-seq_WT_rep2.bed
KAS-seq_KO_rep1.bed
KAS-seq_KO_rep2.bed ---KAS-seq.txt
-h/--help: print this help and exit.
Note: The 'KAS-Analyzer FRiP' shell script is applied to calculate and plot fraction of reads in peaks (FRiP) scores.
fragmentsize
Measure the fragment size of paired-end KAS-seq data..
Usage: KAS-Analyzer fragmentsize [ -h/--help ] [ -o prefix ] [ -l labels ] [ -k KAS-seq ]
Example: nohup KAS-Analyzer fragmentsize -o KAS-seq_fragmentsize -l labels.txt -k KAS-seq.txt &
-o [prefix]: please specify the prefix (basename) of 'KAS-Analyzer fragmentsize' output files. REQUIRED.
-l [labels.txt]: please input the text file containing the labels of paired-end KAS-seq or spKAS-seq data that show in fragment size plot. DEFAULT: basename of (sp)KAS-seq bed files.
Example:
WT.rep1
WT.rep2
WT.rep3
WT.rep4 ---labels.txt
-k [KAS-seq.txt]: please input the text file containing bed files (uniquely mapped reads from 'KAS-Analyzer (sp)KAS-seq'), which are used to calcuate fragment size of DNA fragments. The order and number of (sp)KAS-seq bed files should be the consistent with the labels in labels.txt file. REQUIRED.
Example:
KAS-seq_WT_PE.rep1.bed
KAS-seq_WT_PE.rep2.bed
KAS-seq_WT_PE.rep3.bed
KAS-seq_WT_PE.rep4.bed ---KAS-seq.txt
-h/--help: print this help and exit.
Note: The 'KAS-Analyzer fragmentsize' shell script is applied to calculate and plot fragment size of (sp)KAS-seq data. Note: it only works for paired-end (sp)KAS-seq data.
correlation
Calculate the correlation coefficient and pvalue, generate correlation plot for replicates of KAS-seq data.
Usage: KAS-Analyzer correlation [ -h/--help ] [ -m correlation method ] [ -t threads ] [ -s assembly id ] [ -r regions ] [ -f peaks file ] [ -p plot types ] [ -o prefix ] [ -l labels ] [ -k KAS-seq ]
Example:
On peaks:
nohup KAS-Analyzer correlation -m pearson -t 10 -s hg19 -r peaks -f KAS-seq_peaks.bed -p heatmap -o KAS-seq -l labels.txt -k KAS-seq.txt &
On bins:
nohup KAS-Analyzer correlation -m pearson -t 10 -s hg19 -r bins -p heatmap -o KAS-seq -l labels.txt -k KAS-seq.txt &
-m [correlation method]: please specify the methods to calculate correlation coefficients. e.g. pearson, kendall or spearman. DEFAULT: pearson.
-t [threads]: please specify the number of threads. DEFAULT: 1.
-s [assembly id]: please specify the genome assembly id of your (sp)KAS-seq data. e.g. Human: hg18, hg19, hg38; Mouse: mm9, mm10, mm39; C.elegans: ce10, ce11; D.melanogaster: dm3, dm6; Rat: rn6, rn7; Zebra fish: danRer10, danRer11. REQUIRED.
-r [regions]: please specify the region types to calculate the (sp)KAS-seq density matrix. e.g. bin or peak. DEFAULT: bin.
-f [peaks file]: please input the merged peaks list file. Note: only valid when '-r peaks' is specified. REQUIRED.
-p [plot types]: please specify the plot types to generate correlation plot. DEFAULT: scatterplot.
-o [prefix]: please input the prefix (basename) of 'KAS-Analyzer correlation' output files. REQUIRED.
-l [labels]: please input the text file containing the labels of (sp)KAS-seq data that show in heatmap or scatterplot. REQUIRED.
Example:
WT_rep1
WT_rep2
WT_rep3
KO_rep1
KO_rep2
KO_rep2 ---labels.txt
-k [KAS-seq]: please input the text file containing the indexed bam files of (sp)KAS-seq data that used to calculate correlation coefficient and generate correlation plots. REQUIRED.
Example:
KAS-seq_WT.rep1.bam
KAS-seq_WT.rep2.bam
KAS-seq_WT.rep3.bam
KAS-seq_KO.rep1.bam
KAS-seq_KO.rep2.bam
KAS-seq_KO.rep3.bam ---KAS-seq_data.txt
-h: print this help and exit.
Note: The 'KAS-Analyzer correlation' shell script is applied to calculate correlation coefficients and generate correlation plots between replicates or (sp)KAS-seq data of different conditions.
saturation
Perform saturation analysis for KAS-seq data.
Usage: KAS-Analyzer saturation [ -h/--help ] [ -o prefix ] [ -s assembly id ] [ -c control ] [ -k KAS-seq ]
Example: nohup KAS-Analyzer saturation -o KAS-seq_saturation -c KAS-seq_Input.bed -k KAS-seq.bed &
-o [prefix]: please input the prefix (basename) of 'KAS-Analyzer saturation' output files. DEFAULT: basename of KAS-seq data.
-s [assembly id]: please specify the genome assembly id of (sp)KAS-seq data. e.g. Human: hg18, hg19, hg38; Mouse: mm9, mm10, mm39; C.elegans: ce10, ce11; D.melanogaster: dm3, dm6; Rat: rn6, rn7; Zebra fish: danRer10, danRer11. REQUIRED.
-c [control]: please input the control data (input of (sp)KAS-seq data) containing uniquely mapped reads. e.g. -c KAS-seq_Input.bed. REQUIRED. Note: reads number of KAS-seq and Input should be similar.
-k [KAS-seq]: please input the KAS-seq data containing uniquely mapped reads. e.g. -k KAS-seq.bed. REQUIRED.
-h/--help: print this help and exit.
Note: The 'KAS-Analyzer saturation' shell script is applied to evaluate the saturation of (sp)KAS-seq data.
complexity
Calculate the complexity metric for (sp)KAS-seq data, including the PCR Bottlenecking Coefficient and Non-Redundant Fraction (NRF).
Usage: KAS-Analyzer complexity [ -h/--help ] [ -o prefix ] [ -l labels ] [ -k KAS-seq ]
Example: nohup KAS-Analyzer complexity -o KAS-seq_complexity -l labels.txt -k KAS-seq.txt &
-o [KAS-seq_complexity]: please input the prefix (basename) of 'KAS-Analyzer complexity' output files. REQUIRED.
-l [labels.txt]: please input the text file containing the labels of KAS-seq or spKAS-seq data complexity metric. DEFAULT: basename of KAS-seq files.
Example:
WT.rep1
WT.rep2
WT.rep3
WT.rep4 ---labels.txt
-k [KAS-seq.txt]: please input the text file containing the bam files without removing redundant reads, which are used to calculate the complexity metric including PCR Bottlenecking Coefficients (PBC) and Non-Redundant Fraction (NRF). The order and number of KAS-seq data should be the consistent with the labels file. REQUIRED.
Example:
KAS-seq.rep1.bam
KAS-seq.rep2.bam
KAS-seq.rep3.bam
KAS-seq.rep4.bam ---KAS-seq.txt
-h/--help: print this help and exit.
Note: The 'KAS-Analyzer complexity' shell script is applied to calculate the library complexity metric of (sp)KAS-seq data including, the PCR Bottlenecking Coefficient and Non-Redundant Fraction (NRF) for KAS-seq. Please refer to https://www.encodeproject.org/data-standards/terms/ for more details about the library complexity metric.
genomicdist
Visualize the genomic distribution for KAS-seq peaks (table and plot).
Usage: KAS-Analyzer genomicdist [ -h/--help ] [ -o prefix ] [ -c ] [ -p peaks ] [ -s assembly id ]
Example: nohup KAS-Analyzer genomicdist -o KAS-seq_genomic_distribution -p KAS-seq_peaks.bed -s hg19 &
-o [prefix]: please input the prefix (basename) of 'KAS-Analyzer genomicdist' output files. DEFAULT: basename of KAS-seq peak file.
-c: please specify if the percentages of normal genomic feature distribution is generated, which is regard as a control. DEFAULT: off.
-p [peaks]: please input the KAS-seq peak or differential KAS-seq peak file. REQUIRED.
-s [assembly id]: please specify the reference genome assembly id, e.g. Human: hg18, hg19, hg38; Mouse: mm9, mm10, mm39; C.elegans: ce10, ce11; D.melanogaster: dm3, dm6; Rat: rn6, rn7; Zebra fish: danRer10, danRer11. Note: the assembly id need to be consistent with the reference genome index. REQUIRED.
-h/--help: print this help and exit.
Note: The 'KAS-Analyzer genomicdist' shell script is applied to calculate and plot the percentages of (sp)KAS-seq peaks distribution on genomic features (Promoter(TSS +/-1kb), Exon, Intron, Terminal(TES+3kb) and Intergenic regions).
fingerprint
Plot fingerprint for KAS-seq data.
Usage: KAS-Analyzer fingerprint [ -h/--help ] [ -t threads ] [ -s assembly id ] [ -o prefix ] [ -l labels ] [ -k KAS-seq ]
Example: nohup KAS-Analyzer fingerprint -t 10 -s hg19 -o KAS-seq_fingerprint -l labels.txt -k KAS-seq_data.txt &
-t [threads]: please input the number of threads used for generating KAS-seq fingerprint plot. DEFAULT: 1.
-s [assembly id]: please specify the reference genome assembly id, e.g. Human: hg18, hg19, hg38; Mouse: mm9, mm10, mm39; C.elegans: ce10, ce11; D.melanogaster: dm3, dm6; Rat: rn6, rn7; Zebra fish: danRer10, danRer11. Note: the assembly id need to be consistent with the reference genome index. REQUIRED.
-o [prefix]: please input the prefix (basename) of 'KAS-Analyzer fingerprint' output files. REQUIRED
-l [labels] please input the text file containing the labels of KAS-seq or spKAS-seq data that show in fingerprint plot. REQUIRED.
Example:
KAS-seq.rep1
KAS-seq.rep2
Input.rep1
Input.rep2 ---labels.txt
-k [KAS-seq]: please input the text file containing the bam files of (sp)KAS-seq data. REQUIRED.
Example:
KAS-seq.rep1.bam
KAS-seq.rep2.bam
KAS-seq_Input.rep1.bam
KAS-seq_Input.rep2.bam ---KAS-seq.txt
-h/--help: print this help and exit.
Note: the 'KAS-Analyzer fingerprint' shell script is applied to generate the fingerprint plot of (sp)KAS-seq data. For the more details about fingerprint plot, please refer to https://deeptools.readthedocs.io/en/develop/content/tools/plotFingerprint.html.
Map sub-commands:
trim
Trim adapter and low quality sequence, perform quality control for raw KAS-seq data.
Usage: KAS-Analyzer trim [ -h ] [ -a adapter ] [ -t threads ] [ -f ] [ -q quality ] [ -l length ] [ -1 read1 ] [ -2 read2 ]
Example:
Single-end:
nohup KAS-Analyzer trim -a illumina -t 10 -1 KAS-seq.fastq.gz &
Paired-end:
nohup KAS-Analyzer trim -a illumina -t 10 -1 KAS-seq.R1.fastq.gz -2 KAS-seq.R2.fastq.gz &
-a [adapter types]: adapter sequence to be trimmed. e.g. illumina, nextera or small_rna. Hint: most of the NGS data used Illumina adapter. If not specified explicitly. KAS-Analyzer trim will auto-detect.
-t [threads]: number of threads to be used for trimming. DEFAULT: 1.
-f: instruct 'KAS-Analyzer trim' to check quality control before trimming. DEFAULT: off.
-q [quality]: trim low-quality ends from reads in addition to adapter removal. DEFAULT Phred score(ASCII+33): 20.
-l [length]: discard reads that became shorter than length INT bp because of either quality or adapter trimming. DEFAULT: 30.
-1 [read1]: please input single-end KAS-seq raw fastq file or read 1 of paired-end KAS-seq raw fastq files. REQUIRED.
-2 [read2]: please input read2 of paired-end KAS-seq raw fastq files.
-h: print this help and exit.
Note: The 'KAS-Analyzer trim' shell script mainly invoke the trim-galore, please refer to http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ for more information.
KAS-seq
Align KAS-seq data to the reference genome, deduplicate mapped reads, and generate several files with maped reads (bam, bed and bedGraph).
Usage: KAS-Analyzer KAS-seq [ -h ] [ -a aligner ] [ -t threads ] [ -i index path ] [ -u ] [ -e extend length ] [ -o prefix ] [ -s assembly id ] [ -1 read1 ] [ -2 read2 ]
Example:
Single-end:
nohup KAS-Analyzer KAS-seq -a bowtie2 -t 10 -i /absolute path/hg19_Bowtie2Index/hg19 -e 150 -o KAS-seq -s hg19 -1 KAS-seq.trim.fastq.gz &
Paired-end:
nohup KAS-Analyzer KAS-seq -a bowtie2 -t 10 -i /absolute path/hg19_Bowtie2Index/hg19 -o KAS-seq -s hg19 -1 KAS-seq.trim.R1.fastq.gz -2 KAS-seq.trim.R2.fastq.gz &
Note: Bowtie2 Index example: /absolute path/Bowtie2Index/hg19; bwa Index example: /absolute path/BWAIndex/hg19.fa
-a [aligner]: please specify the aligner (bowtie2 or bwa) you want to use to map KAS-seq data. DEFAULT: bowtie2.
-t [threads]: please input the number of threads used for KAS-seq data mapping. DEFAULT: 1.
-i [index path]: please input the absolute path of reference genome index for aligner. REQUIRED.
-u: please specify to filter the unique mapped reads. DEFAULT: off.
-e [extendlengthHelp]: please input the extend length for single-end KAS-seq data. DEFAULT: 150.
-o [prefix]: please input the prefix (basename) of 'KAS-Analyzer KAS-seq' output files. REQUIRED.
-s [assembly id]: please specify the reference genome assembly id, e.g. Human: hg18, hg19, hg38; Mouse: mm9, mm10, mm39; C.elegans: ce10, ce11; D.melanogaster: dm3, dm6; Rat: rn6, rn7; Zebra fish: danRer10, danRer11. Note: the assembly id need to be consistent with the reference genome index. REQUIRED.
-1 [read1]: please input trimmed single-end KAS-seq fastq file or read 1 of paired-end KAS-seq raw fastq files; Compressed .fastq.gz is accepted. REQUIRED.
-2 [read2]: please input trimmed read2 of paired-end KAS-seq raw fastq files. Compressed .fastq.gz is accepted.
-h: print this help and exit.
Note: The 'KAS-Analyzer KAS-seq' shell script mainly invoke the specific aligner (bowtie, bowtie2 or bwa) for KAS-seq data mapping, please refer to their official websites for more information.
spKAS-seq
Align strand specific KAS-seq (spKAS-seq) data.
Usage: KAS-Analyzer spKAS-seq [ -h ] [ -t threads ] [ -i index path ] [ -u ] [ -r ] [ -f fold change ] [ -b bin size ] [ -e extend length ] [ -o prefix ] [ -s assembly id ] [ -1 read1 ] [ -2 read2 ]
Note: we strongly recommend paired-end sequencing for strand specific KAS-seq (spKAS-seq) data to accurately measure the fragments size.
Example:
Single-end:
nohup KAS-Analyzer spKAS-seq -t 10 -i /absolute path/hg19_Bowtie2Index/hg19 -o spKAS-seq -r -s hg19 -1 spKAS-seq.trim.R1.fastq.gz &
Paired-end:
nohup KAS-Analyzer spKAS-seq -t 10 -i /absolute path/hg19_Bowtie2Index/hg19 -o spKAS-seq -r -s hg19 -1 spKAS-seq.trim.R1.fastq.gz -2 spKAS-seq.trim.R2.fastq.gz &
Note: Bowtie2 Index example: /absolute path/Bowtie2Index/hg19.
-t [threads]: please specify the number of threads used for spKAS-seq data mapping. DEFAULT: 1.
-i [index path]: please input the absolute path of reference genome index for aligner. REQUIRED.
-u: please specify to filter the unique mapped reads. DEFAULT: off.
-r: please specify if identify R-loops regions with spKAS-seq data. DEFAULT: off.
-f [fold change cutoff]: please specify the fold change cutoff of spKAS-seq reads difference between plus and minus strands used for R-loops identification. DEFAULT: 2.
-b [bin size]: please specify the size of bins used to identify R-loops. DEFAULT: 500.
-o [prefix]: please input the prefix (basename) of 'KAS-Analyzer spKAS-seq' output files. REQUIRED.
-s [assembly id]: please input the reference genome assembly id, e.g. Human: hg18, hg19, hg38; Mouse: mm9, mm10, mm39; C.elegans: ce10, ce11; D.melanogaster: dm3, dm6; Rat: rn6, rn7; Zebra fish: danRer10, danRer11. Note: the assembly id need to be consistent with the reference genome index. REQUIRED.
-1 [read1]: please input trimmed single-end spKAS-seq fastq file or read1 of paired-end spKAS-seq fastq files; compressed .fastq.gz is accepted. REQUIRED.
-2 [read2]: please input trimmed read2 of paired-end spKAS-seq raw fastq files. compressed fastq.gz is accepted.
-h: print this help and exit.
Note: The 'KAS-Analyzer spKAS-seq' shell script mainly invoke bowtie2 for spKAS-seq data mapping and R-loops identification, please refer to their official websites for more information.
peakscalling
Call broad or sharp peaks for KAS-seq data.
Usage: KAS-Analyzer peakscalling [ -h ] [ -m peaks caller ] [ -t KAS-seq ] [ -c Control ] [ -b mode ] [ -o prefix ] [ -p pvalue or qvalue ] [ -g assembly id ]
Example: nohup KAS-Analyzer peakscalling -t KAS-seq.rep1.bed,KAS-seq.rep2.bed -c Control_Input.rep1.bed,Control_Input.rep2.bed -o KAS-seq -g hg19 &
-m [peaks caller]: please input the peaks caller (macs2, epic2 or macs2_and_epic2) that you want to use for KAS-seq peaks calling. DEFAULT: macs2_and_epic2.
-t [KAS-seq]: please input the KAS-seq bed or bam files. e.g. KAS-seq.rep1.bed,KAS-seq.rep2.bed or KAS-seq.rep1.bam,KAS-seq.rep2.bam. REQUIRED.
-c [Control]: please input the KAS-seq control bed or bam files. e.g. KAS-seq_Input.rep1.bed,KAS-seq_Input.rep2.bed or KAS-seq_Input.rep1.bam,KAS-seq_Input.rep2.bam. OPTIONAL.
-b [mode]: specify macs2 to perferm KAS-seq peaks calling with 'broad', 'sharp' or 'both' mode. epic2 for broad peaks; macs2 for sharp peaks; combined epic2&macs2 for broad and sharp peaks . DEFAULT: both.
-o [prefix]: please input the prefix (basename), which will be used to generate the name of 'KAS-Analyzer peakscalling' output files. REQUIRED.
-p [FDR or qvalue]: please input the pvalue or qvalue for KAS-seq peaks calling with macs14 or macs2. DEFAULT: epic2 FDR: 0.05; macs2 qvalue: 0.01
-g [assembly id]: please specify the reference genome assembly id of KAS-seq data. e.g. Human: hg18, hg19, hg38; Mouse: mm9, mm10, mm39; C.elegans: ce10, ce11; D.melanogaster: dm3, dm6; Rat: rn6, rn7; Zebra fish: danRer10, danRer11. REQUIRED.
-h: print this help and exit.
Note: This shell script mainly invoke macs2 and epic2 for calling (sp)KAS-seq data peaks, please google their github pages for more information.
normalize
Normalize KAS-seq data with bedGraph density files.
Usage: KAS-Analyzer normalize [ -h/--help ] [-m methods ] [ -k KAS-seq ] [ -r ratios ] [ -b ] [ -s assembly id ]
Example: nohup KAS-Analyzer normalize -m ratios -k KAS-seq_data.txt -r ratios.txt -b -s mm10 &
-m [methods]: please input the methods used for KAS-seq data normalization. e.g. ratios or RPKM. DEFAULT: ratios.
-k [KAS-seq_data.txt]: please input the text file containing the bedGraph files generated from 'KAS-Analyzer (sp)KAS-seq'. REQUIRED.
Example:
-m ratios: -m FPKM:
KAS-seq_WT.rep1.bg KAS-seq_WT.rep1.bam
KAS-seq_WT.rep2.bg KAS-seq_WT.rep2.bam
KAS-seq_KO.rep1.bg KAS-seq_KO.rep1.bam
KAS-seq_KO.rep2.bg ---KAS-seq_data.txt KAS-seq_KO.rep1.bam ---KAS-seq_data.txt
-r [ratios.txt]: please input the text file containing ratios that used to normalize KAS-seq data, which can be calculated based on mapped reads number or SpikeIn reads. The order and number of ratios should be the consistent with KAS-seq bedGraph files. REQUIRED.
Example:
1.10
1.20
1.30
1.23 ---ratios.txt
-b: please specify if you want to convert the normalized bedGraph files into bigWig files. DEFAULT: off.
-s [assembly id]: please input the reference genome assembly id of bedGraph files. e.g. Human: hg18, hg19, hg38; Mouse: mm9, mm10, mm39; C.elegans: ce10, ce11; D.melanogaster: dm3, dm6; Rat: rn6, rn7; Zebra fish: danRer10, danRer11. Note: the assembly id need to be consistent with the normalized KAS-seq bedGraph files. REQUIRED only if -b was specified.
-h/--help: print this help and exit.
Note: The 'KAS-Analyzer normalize' shell script is applied to normalize spKAS-seq or KAS-seq data.
ToBigWig
Convert normalized bedGraph file to bigWig file.
Usage: KAS-Analyzer ToBigWig [ -h/--help ] [ -k KAS-seq ] [ -s assembly id ]
Example: nohup KAS-Analyzer ToBigWig -k KAS-seq_data.txt -s hg19 &
-k [KAS-seq]: please input the text file containing the bedGraph files generated from 'KAS-Analyzer KAS-seq'. REQUIRED.
Example:
KAS-seq_WT.rep1.nor.bg
KAS-seq_WT.rep2.nor.bg
KAS-seq_KO.rep1.nor.bg
KAS-seq_KO.rep2.nor.bg ---KAS-seq_data.txt
-s [assembly id]: please input the reference genome assembly id, e.g. Human: hg18, hg19, hg38; Mouse: mm9, mm10, mm39; C.elegans: ce10, ce11; D.melanogaster: dm3, dm6; Rat: rn6, rn7; Zebra fish: danRer10, danRer11. Note: the assembly id need to be consistent with the normalized KAS-seq bedGraph files. REQUIRED.
-h/--help: print this help and exit.
Note: The 'KAS-Analyzer ToBigWig' shell script is applied to transfer (sp)KAS-seq bedGraph to bigWig files.
targetgenes
Define target or associated genes (promoter, genebody, terminator or gene) of KAS-seq peaks, R-loops or enhancers loci.
Usage: KAS-Analyzer targetgenes [ -h/--help ] [ -o prefix ] [ -s assembly id ] [ -f features ] [ -l length ] [ -p peaks ]
Example:
Gene features:
nohup KAS-Analyzer targetgenes -o KAS-seq_peaks_target_genes -s mm10 -f promoter -p KAS-seq_peaks.bed &
Associated genes of enhancers:
nohup KAS-Analyzer targetgenes -o KAS-seq_ss_enhancers_asso_genes -s mm10 -f enhancer -l 50000 -p KAS-seq_ss_enhancers.bed &
-o [prefix]: please input the prefix (basename) of 'KAS-Analyzer targetgenes' output files. DEFAULT: basename of peaks file.
-s [assembly id]: please specify the genome assembly id of KAS-seq peaks. -s [assembly id]. e.g. Human: hg18, hg19, hg38; Mouse: mm9, mm10, mm39; C.elegans: ce10, ce11; D.melanogaster: dm3, dm6; Rat: rn6, rn7; Zebra fish: danRer10, danRer11. REQUIRED.
-f [features]: please specify the gene feagures used to define target genes. e.g. promoter, genebody, terminator, gene or enhancer. REQUIRED.
-l [length]: please specify the length cutoff (length to TSS) to define enhancer's associated genes. DEFAULT: 50000.
-p [peaks]: please specify the KAS-seq peaks, R-loops or enhancer to define their target genes. REQUIRED.
-h/--help: print this help and exit.
Note: The 'KAS-Analyzer targetgenes' shell script is applied to define target or associated genes (promoter, genebody, terminator or gene) of KAS-seq peaks, R-loops or enhancers loci.
UCSC
Generate bedGraph files ready for submitting to UCSC genome browser.
Usage: KAS-Analyzer UCSC [ -h/--help ] [ -k KAS-seq ] [ -l UCSC track ] [ -c track colors ]
Example: nohup KAS-Analyzer UCSC -k KAS-seq_data.txt -n UCSC_track_names.txt &
-k [KAS-seq]: please input the text file containing the bedGraph files generated from 'KAS-Analyzer KAS-seq'. REQUIRED.
Example:
KAS-seq_WT.rep1.nor.bg
KAS-seq_WT.rep2.nor.bg
KAS-seq_KO.rep1.nor.bg
KAS-seq_KO.rep2.nor.bg ---KAS-seq_data.txt
-l [UCSC track]: please input the text file containing the track names of KAS-seq or spKAS-seq data that you want to visualize on UCSC genome browser. REQUIRED.
Example:
KAS-seq_WT.rep1
KAS-seq_WT.rep2
KAS-seq_KO.rep1
KAS-seq_KO.rep2 ---UCSC_track_names.txt
-c [track colors]: please input the text file containing the R,G,B colors list for KAS-seq or spKAS-seq data that you want to visualize on UCSC genome browser. DEFAULT: black (0,0,0).
Example:
255,102,102
255,178,102
102,255,102
102,178,255 ---track_colors.txt
For more colors options, please refer to https://www.w3schools.com/colors/colors_rgb.asp or https://www.rapidtables.com/web/color/RGB_Color.html.
-h/--help: print this help and exit.
Note: The 'KAS-Analyzer UCSC' shell script is used to generate files for uploading into UCSC genome browser.
Summary Plot sub-commands:
profile
Generate metagene profile for KAS-seq data.
Usage: KAS-Analyzer profile [ -h/--help ] [ -t threads ] [ -s assembly id ] [ -e length ] [ -o prefix ] [ -r regions ] [ -p peaks file ] [ -f peaks files list ] [ -l labels ] [ -c colors ] [ -k KAS-seq ]
Example:
Genomic features(genebody, TSS or TES):
nohup KAS-Analyzer profile -t 10 -s hg19 -o KAS-seq_genebody -r genebody -c red,blue,green,purple -l labels.txt -k KAS-seq.txt &
Custom regions(peaks. e.g. enhancers.bed):
nohup KAS-Analyzer profile -t 10 -o KAS-seq_peaks -r peaks -p KAS-seq_peaks.bed -c red,blue,green,purple -l labels.txt -k KAS-seq.txt &
KAS-seq signal on different regions (-f [peaks list] must be specified):
nohup KAS-Analyzer profile -t 10 -o KAS-seq_different_clusters -r peakslist -f peaks_cluster1.bed,peaks_cluster2.bed,peaks_cluster3.bed -c red,green,purple -l labels.txt -k KAS-seq.txt &
-t [threads]: please input the number of threads used for generating (sp)KAS-seq metagene profile plot. DEFAULT: 1.
-s [assemblyid]: please specify the reference genome assembly id, e.g. Human: hg18, hg19, hg38; Mouse: mm9, mm10, mm39; C.elegans: ce10, ce11; D.melanogaster: dm3, dm6; Rat: rn6, rn7; Zebra fish: danRer10, danRer11. Note: the assembly id need to be consistent with the reference genome index. REQUIRED only for 'genomic features' mode.
-o [KAS-seq_profile]: please input the prefix (basename), which will be used to generate the name of 'KAS-Analyzer metageneprofile' output files. REQUIRED.
-e [length]: please specify the distance upstream of the start site of the regions defined in the region file. If the regions are genebody, this would be the distance upstream of the transcription start site. DEFAULT: 3000.
-r [regions]: please specify the regions types for generating metagene profile plot. e.g. Genomic features: genebody, TSS or TES; Custom regions: peaks or peakslist. REQUIRED.
-p [peaks file]: please specify the peak file. REQUIRED only for 'custom regions: peaks' mode.
-f [peaks list]: please specify the peak files list. e.g. peaks_cluster1.bed,peaks_cluster2.bed,peaks_cluster3.bed. REQUIRED only for 'custom regions: peakslist' mode. Note: only one KAS-seq bigWig file is needed.
-c [colors]: please specify the color list for (sp)KAS-seq data in metagene profile plot. Note: the number of colors in the profile plot needs to be consistent with the number of KAS-seq bigWig files. REQUIRED. Note: the list of valid color names https://matplotlib.org/examples/color/named_colors.html.
-l [labels.txt]: please input the text file containing the labels of (sp)KAS-seq data or peaks files (-f need to be specified) that used for generating metagene profile. DEFAULT: basename of (sp)KAS-seq bigWig files or peaks files.
Example:
WT_rep1 Cluster1
WT.rep2 Cluster2
KO.rep1 Cluster3
KO.rep2 or ---labels.txt
-k [KAS-seq.txt]: please input the text file containing normalized (sp)KAS-seq bigWig files, which can be generated with 'KAS-Analyzer normalize' and 'KAS-Analyzer bedGraphToBigWig' shell scripts. The order and number of (sp)KAS-seq bigWig files should be the consistent with the labels file when provided. REQUIRED.
Example:
KAS-seq_WT_rep1.nor.bigWig
KAS-seq_WT_rep2.nor.bigWig
KAS-seq_KO_rep1.nor.bigWig
KAS-seq_KO_rep2.nor.bigWig ---KAS-seq.txt
-h/--help: print this help and exit.
Note: The 'KAS-Analyzer profile' shell script is applied to generate metagene profile for (sp)KAS-seq data on genomic features( genebody, TSS or TES) or provided custom regions. 'KAS-Analyzer metageneprofile' shell script mainly invoke deeptools 'computeMatrix' and 'plotProfile', please refer to https://deeptools.readthedocs.io/en/develop/content/list_of_tools.html for more information.
heatmap
Generate heatmap for (sp)KAS-seq data.
Usage: KAS-Analyzer heatmap [ -h/--help ] [ -t threads ] [ -e length ] [ -s assembly id ] [ -q ] [ -u samples using ] [ -m maximum value ] [ -o prefix ] [ -r regions ] [ -p peaks ] [ -l labels ] [ -c colors ] [ -k KAS-seq ]
Example:
Genomic features(genebody, TSS or TES):
nohup KAS-Analyzer heatmap -t 10 -s hg19 -o KAS-seq_heatmap -r genebody -q -c Reds -l labels.txt -k KAS-seq.txt &
Custom regions(peaks. e.g. enhancers.bed):
nohup KAS-Analyzer heatmap -t 10 -o KAS-seq_heatmap -r peaks -q -p KAS-seq_peaks.bed -c Reds,Reds,Blues,Blues -l labels.txt -k KAS-seq.txt &
-t [threads]: please specify the number of threads used for generating (sp)KAS-seq heatmap plot. DEFAULT: 1.
-s [assemblyid]: please specify the genome assembly id, e.g. Human: hg18, hg19, hg38; Mouse: mm9, mm10, mm39; C.elegans: ce10, ce11; D.melanogaster: dm3, dm6; Rat: rn6, rn7; Zebra fish: danRer10, danRer11. REQUIRED only for 'genomic features' mode.
-q: please specify to sort the regions. DEFAULT: off.
-u [sample using]: please specify the samples list to sort the regions. e.g. -u 1,2,3
-e [length]: please specify the distance upstream of the start site of the regions defined in the region file. If the regions are genebody, this would be the distance upstream of the transcription start site. DEFAULT: 3000.
-m [maximum value]: please specify the maximum value of the heatmap intensities. e.g. -m 15,20,60. Note: the number of values need to be consistent with KAS-seq samples.
-o [KAS-seq_heatmap]: please specify the prefix (basename) of 'KAS-Analyzer heatmap' output files. REQUIRED.
-r [regions]: please specify the regions types to generate heatmap plot. e.g. Genomic features: genebody, TSS or TES; Custom regions: peaks. REQUIRED.
-p [peakslist]: please input the peak list file. REQUIRED only for 'custom regions' mode.
-c [colors]: please specify the colors for (sp)KAS-seq data in heatmap plot. Note: you can specify only one colors (e.g. Reds) or specify a color list (e.g. Reds,Greens,Blues,Purples), which needs to be consistent with the number of KAS-seq bigWig files. REQUIRED. Note: please refer to http://matplotlib.org/users/colormaps.html to get the list of valid colors names.
-l [labels.txt]: please input the text file containing the labels of (sp)KAS-seq data to generate metagene profile. Default: basename of (sp)KAS-seq bigWig files.
Example:
WT_rep1
WT.rep2
KO.rep1
KO.rep2 ---labels.txt
-k [KAS-seq.txt]: please input the text file containing normalized (sp)KAS-seq bigWig files, which can be generated with 'KAS-Analyzer normalize' and 'KAS-Analyzer bedGraphToBigWig' shell scripts. The order and number of (sp)KAS-seq bigWig files should be the consistent with the labels file when specified. REQUIRED.
Example:
KAS-seq_WT_rep1.nor.bigWig
KAS-seq_WT_rep2.nor.bigWig
KAS-seq_KO_rep1.nor.bigWig
KAS-seq_KO_rep2.nor.bigWig ---KAS-seq.txt
-h/--help: print this help and exit.
Note: The 'KAS-Analyzer heatmap' shell script is applied to generate heatmap plots for (sp)KAS-seq data on genomic features( genebody, TSS or TES) or provided custom regions. 'KAS-Analyzer heatmap' shell script mainly invoke deeptools 'computeMatrix' and 'plotHeatmap', please refer to https://deeptools.readthedocs.io/en/develop/content/list_of_tools.html for more information.
Differential analysis sub-commands:
diff
Perform differential KAS-seq analysis on promoter, genebody, gene, bin or custom regions.
Usage: KAS-Analyzer diff [ -h/--help ] [ -t threads ] [ -o prefix ] [ -s assembly id ] [ -r regions ] [ -p peaks ] [ -f fold change ] [ -c comparison file ] [ -l labels ] [ -k KAS-seq ]
Example: nohup KAS-Analyzer diff -o KAS-seq_diff -t 10 -s mm10 -r gene -c comparision.txt -l labels.txt -k KAS-seq_data.txt &
-t [threads]: please specify the number of threads used for calculating KAS index. DEFAULT: 1.
-o [prefix]: please input the prefix (basename) of 'KAS-Analyzer diff' output files. DEFAULT: basename of KAS-seq txt file.
-s [assembly id]: please specify the genome assembly id of KAS-seq data. -s [assembly id]. e.g. Human: hg18, hg19, hg38; Mouse: mm9, mm10, mm39; C.elegans: ce10, ce
11; D.melanogaster: dm3, dm6; Rat: rn6, rn7; Zebra fish: danRer10, danRer11. REQUIRED.
-r [regions]: please specify the types of genomic regions used for differential KAS-seq analysis. e.g. promoter, genebody, gene, bin or peak. REQUIRED.
-p [peaks]: please specify the custom regions file used for differential KAS-seq analysis, if regions type is set as 'peak'. OPTIONAL.
-f [fold change]: please specify the fold change used for identify regions with differential KAS-seq density. DEFAULT: 2.
-c [comparison]: please input the text file containing the comparison information for differential KAS-seq analysis. REQUIRED.
condition
WT.rep1 WT
WT.rep2 WT
KO.rep1 KO
KO.rep2 KO ---comparison.txt
-l [labels]: please input the text file containing the labels of (sp)KAS-seq data that show in output files, which need to be consistent with comparision file. REQUIRED.
Example:
WT.rep1
WT.rep2
KO.rep1
KO.rep2 ---labels.txt
-k [KAS-seq]: please input the text file containing the bigWig files of (sp)KAS-seq data that used for differential KAS-seq analysis. REQUIRED.
Example:
KAS-seq_WT.rep1.bigWig
KAS-seq_WT.rep2.bigWig
KAS-seq_KO.rep1.bigWig
KAS-seq_KO.rep2.bigWig ---KAS-seq_data.txt
-h/--help: print this help and exit.
Note: The 'KAS-Analyzer diff' shell script is applied to perform differential KAS-seq analysis on promoter, genebody, gene, bin or custom regions.
TC
Perform ‘case-only’ or ‘case-control’ differential time course(TC) analysis for (sp)KAS-seq data.
Usage: KAS-Analyzer TC [ -h/--help ] [ -t threads ] [ -n ratios ] [ -o prefix ] [ -g assembly id ] [ -r regions ] [ -s bin size ] [ -b ] [ -p peaks ] [ -d differential analysis ] [ -a annotation ] [ -l labels ] [ -k KAS-seq ]
Example:
Case-only:
nohup KAS-Analyzer TC -o KAS-seq_timecourse -t 10 -g mm10 -r bin -d case_only -a Case_only.annotation.txt -l labels.txt -k KAS-seq_data.txt &
Case-control:
nohup KAS-Analyzer TC -o KAS-seq_timecourse -t 10 -g mm10 -r bin -d case_control -a Case_control.annotation.txt -l labels.txt -k KAS-seq_data.txt &
-t [threads]: please specify the number of threads used for calculating KAS index. DEFAULT: 1.
-n [ratios.txt]: please input the text file containing ratios that used to normalize KAS-seq data undergo global significant changes, which can be calculated based on SpikeIn reads. The order and number of ratios should be the consistent with KAS-seq bam files. OPTIONAL.
Example:
1.10
1.20
1.30
1.23 ---ratios.txt
-o [prefix]: please input the prefix (basename) of 'KAS-Analyzer KASindex' output files. REQUIRED.
-g [assembly id]: please specify the genome assembly id of KAS-seq data. -g [assembly id]. e.g. Human: hg18, hg19, hg38; Mouse: mm9, mm10, mm39; C.elegans: ce10, ce11; D.melanogaster: dm3, dm6; Rat: rn6, rn7; Zebra fish: danRer10, danRer11. REQUIRED.
-r [regions]: please specify the types of genomic regions. e.g. promoter, genebody, bin, gene or peak. REQUIRED.
-s [bin size]: please specify the size of bins if -r is set to 'bin'. DEFAULT:1000.
-b: please specify if KAS-seq data have batch effects. DEFAULT: off.
-p [peaks]: please specify the custom regions file for perform differential time course KAS-seq analysis. OPTIONAL.
-d [diff analysis type]: please specify the types of differential time course KAS-seq analysis. e.g. case_only or case_control. REQUIRED.
-a [annotation]: please specify the annotation file. REQUIRED. Example:
1) Case-only: or 2) Case-Control:
Sample Condition Time Batch Sample Condition Time Batch or Batch
rep1.0min rep1.0min case 1 B_NULL WT.rep1.0min WT.rep1.0min control 1 B_NULL B1
rep2.0min rep2.0min case 1 B_NULL WT.rep2.0min WT.rep2.0min control 1 B_NULL B2
rep3.0min rep3.0min case 1 B_NULL WT.rep3.0min WT.rep3.0min control 1 B_NULL B3
rep1.15min rep1.15min case 2 B_NULL WT.rep1.15min WT.rep1.15min control 2 B_NULL B1
rep2.15min rep2.15min case 2 B_NULL WT.rep2.15min WT.rep2.15min control 2 B_NULL B2
rep3.15min rep3.15min case 2 B_NULL WT.rep3.15min WT.rep3.15min control 2 B_NULL B3
rep1.30min rep1.30min case 3 B_NULL WT.rep1.30min WT.rep1.30min control 3 B_NULL B1
rep2.30min rep2.30min case 3 B_NULL WT.rep2.30min WT.rep2.30min control 3 B_NULL B2
rep3.30min rep3.30min case 3 B_NULL WT.rep3.30min WT.rep3.30min control 3 B_NULL B3
rep1.60min rep1.60min case 4 B_NULL KO.rep1.0min KO.rep1.0min case 1 B_NULL B1
rep2.60min rep2.60min case 4 B_NULL KO.rep2.0min KO.rep2.0min case 1 B_NULL B2
rep3.60min rep3.60min case 4 B_NULL KO.rep3.0min KO.rep3.0min case 1 B_NULL B3
KO.rep1.15min KO.rep1.15min case 2 B_NULL B1
KO.rep2.15min KO.rep2.15min case 2 B_NULL B2
KO.rep3.15min KO.rep3.15min case 2 B_NULL B3
KO.rep1.30min KO.rep1.30min case 3 B_NULL B1
KO.rep2.30min KO.rep2.30min case 3 B_NULL B2
KO.rep3.30min KO.rep3.30min case 3 B_NULL B3 ---annotation.txt
-l [labels]: please input the text file containing the labels of KAS-seq data for differential time course (TC) analysis. REQUIRED. Example:
1) Case-only: or 2) Case-Control:
rep1.0min WT.rep1.0min
rep2.0min WT.rep2.0min
rep3.0min WT.rep3.0min
rep1.15min WT.rep1.15min
rep2.15min WT.rep2.15min
rep3.15min WT.rep3.15min
rep1.30min WT.rep1.30min
rep2.30min WT.rep2.30min
rep3.30min WT.rep3.30min
rep1.60min KO.rep1.0min
rep2.60min KO.rep2.0min
rep3.60min KO.rep3.0min
KO.rep1.15min
KO.rep2.15min
KO.rep3.15min
KO.rep1.30min
KO.rep2.30min
KO.rep3.30min ---labels.txt
-k [KAS-seq]: please input the text file containing indexed bam files of KAS-seq data for differential time course (TC) KAS-seq analysis. REQUIRED. Example:
1) Case-only: or 2) Case-Control:
treat.rep1.0min.bam WT.treat.rep1.0min.bam
treat.rep2.0min.bam WT.treat.rep2.0min.bam
treat.rep3.0min.bam WT.treat.rep3.0min.bam
treat.rep1.15min.bam WT.treat.rep1.15min.bam
treat.rep2.15min.bam WT.treat.rep2.15min.bam
treat.rep3.15min.bam WT.treat.rep3.15min.bam
treat.rep1.30min.bam WT.treat.rep1.30min.bam
treat.rep2.30min.bam WT.treat.rep2.30min.bam
treat.rep3.30min.bam WT.treat.rep3.30min.bam
treat.rep1.60min.bam KO.treat.rep1.0min.bam
treat.rep2.60min.bam KO.treat.rep2.0min.bam
treat.rep3.60min.bam KO.treat.rep3.0min.bam
KO.treat.rep1.15min.bam
KO.treat.rep2.15min.bam
KO.treat.rep3.15min.bam
KO.treat.rep1.30min.bam
KO.treat.rep2.30min.bam
KO.treat.rep3.30min.bam ---KAS-seq_data.txt
-h/--help: print this help and exit.
Note: The 'KAS-Analyzer TC' shell script is applied to performed differential analysis for 'case only' or 'case-control' time course(TC) KAS-seq on promoter, genebody, bin, genes or custom regions.
PCA
Perform and plot PCA analysis for (sp)KAS-seq data.
Usage: KAS-Analyzer PCA [ -h/--help ] [ -o prefix ] [ -t threads ] [ -r regions ] [ -s assembly id ] [ -b bin size ] [ -p peaks] [ -l labels ] [ -k KAS-seq ]
Example: nohup KAS-Analyzer PCA -o KAS-seq_PCA -r bin -s mm10 -l labels.txt -k KAS-seq.txt &
-o [KAS-seq_PCA]: please input the prefix (basename) of 'KAS-Analyzer PCA' output files. REQUIRED.
-t [threads]: please specify the number of threads used for perform PCA analysis. DEFAULT: 1.
-r [regions]: please specify the regions used to perform PCA analysis. e.g. promoter, genebody, peak or bin. DEFAULT: bin.
-s [assemblyid]: please specify the reference genome assembly id of your (sp)KAS-seq data. e.g. Human: hg18, hg19, hg38; Mouse: mm9, mm10, mm39; C.elegans: ce10, ce11; D.melanogaster: dm3, dm6; Rat: rn6, rn7; Zebra fish: danRer10, danRer11. REQUIRED.
-r [regions]: please specify the regions used to perform PCA analysis. e.g. promoter, genebody, peak or bin. DEFAULT: bin.
-b [bin size]: please specify the bin size for bins mode: '-r bins'. DEFAULT: 10000.
-p [peaks file]: please input the custom regions file that used to perform PCA analysis. REQUIRED in 'peak' mode.
-l [labels.txt]: please input the text file containing the labels of (sp)KAS-seq data that shown in the PCA plot. DEFAULT: basename of KAS-seq data.
Example:
0h
4h
8h
12h
24h ---labels.txt
-k [KAS-seq.txt]: please input the text file containing indexed (sp)KAS-seq bam files. The order and number of (sp)KAS-seq data should be the consistent with the labels file if specified. REQUIRED.
Example:
KAS-seq.0h.bam
KAS-seq.4h.bam
KAS-seq.8h.bam
KAS-seq.12h.bam
KAS-seq.24h.bam --KAS-seq.txt
-h/--help: print this help and exit.
Note: The 'KAS-Analyzer PCA' shell script is applied to perform Principal Component Analysis (PCA) for (sp)KAS-seq data.
R-loops sub-commands:
R-loop
Identify R-loops regions with spKAS-seq data.
Usage: KAS-Analyzer R-loop [ -h/--help ] [ -t threads ] [ -o prefix ] [ -s assembly id ] [ -p peaks ] [ -b bin size ] [ -f fold change ] [ -l labels ] [ -n Input ] [ -k spKAS-seq ]
Example: nohup KAS-Analyzer R-loop -o KAS-seq_R-loops -t 10 -s mm10 -l labels.txt -k KAS-seq.txt &
-t [threads]: please specify the number of threads used for R-loops identification. DEFAULT: 1.
-o [prefix]: please input the prefix (basename) of 'KAS-Analyzer R-loop' output files. Default: basename of txt files containing spKAS-seq data.
-s [assembly id]: please specify the genome assembly id of spKAS-seq data. -s [assembly id]. e.g. Human: hg18, hg19, hg38; Mouse: mm9, mm10, mm39; C.elegans: ce10, ce11; D.melanogaster: dm3, dm6; Rat: rn6, rn7; Zebra fish: danRer10, danRer11.
-p [peaks]: please specify the spKAS-seq peaks file. if not specified, spKAS-seq peaks will be called by using macs2 without spKAS-seq Input. OPTIONAL.
-b [bin size]: please specify the size of bins used to identify R-loops. DEFAULT: 500.
-f [fold change]: please specify the fold change cutoff of spKAS-seq reads difference between plus and minus strands used for R-loops identification. DEFAULT: 2.
-l [labels]: please input the text file containing the labels spKAS-seq data that used to identify R-loops. Default: basename of KAS-seq files.
Example:
rep1
rep2
rep3 ---labels.txt
-n [Input]: please input the text file containing Input bed files for R-loops identification. OPTIONAL.
Example:
Input_rep1.bed
Input_rep2.bed
Input_rep3.bed ---Input.txt
-k [KAS-seq]: please input the text file containing spKAS-seq bed files for R-loops identification. The order and number of spKAS-seq data should be the consistent with the labels file in -l [labels]. REQUIRED.
Example:
spKAS-seq_rep1.bed
spKAS-seq_rep2.bed
spKAS-seq_rep3.bed ---KAS-seq.txt
-h/--help: print this help and exit.
Note: The 'KAS-Analyzer R-loops' shell script is applied to identify R-loops from multiples pKAS-seq data.
RNaseH
Identify R-loops regions sensitive to RNase H treatment.
Usage: KAS-Analyzer RNaseH [ -h/--help ] [ -p threads ] [ -o prefix ] [ -r WT_R-loop.bed ] [ -f fold change ] [ -c WT_R-loop_density ] [ -t RNaseH_R-loop_density ]
Example: nohup KAS-Analyzer RNaseH -p 10 -o RNaseH_sensitive_R-loops -r WT_R-loop.bed -f 2 -c WT_R-loop_density.txt -t RNaseH_R-loop_density.txt &
-p [threads]: please specify the number of threads used for R-loops identification. DEFAULT: 1.
-o [prefix]: please input the prefix (basename) of 'KAS-Analyzer RNaseH' output files. REQUIRED.
-r [WT_R-loops.bed]: please specify the R-loop list defined using spKAS-seq data in cells without RNase H treatment. REQUIRED.
-f [fold change]: please specify the fold change threshold of R-loop density difference between WT and RNase H samples used for the identification of R-loops sensitive to RNase H treatment. DEFAULT: 2.
-c [WT_R-loop_density.txt]: please input the text file containing WT R-loop density files. REQUIRED.
Example:
WT_R-loop.rep1.bigWig
WT_R-loop.rep2.bigWig ---WT_R-loop_density.txt
-t [RNaseH_R-loop_density.txt]: please input the text file containing RNase H R-loop density files. REQUIRED.
Example:
RNaseH_R-loop.rep1.bigWig
RNaseH_R-loop.rep2.bigWig ---RNaseH_R-loop_density.txt
-h/--help: print this help and exit.
Note: The 'KAS-Analyzer RNaseH' shell script is applied to identify R-loops sensitive to RNase H treatment.
Single-stranded transcribing enhancers (SST enhancers) identification sub-commands:
SST_enhancer
Identify the single-stranded transcribing enhancers (SST enhancers).
Usage: KAS-Analyzer SST_enhancer [ -h/--help ] [ -o prefix ] [ -t threads ] [ -s assembly id ] [ -e enhancer ] [ -p peaks ] [ -k KAS-seq ]
Example: nohup KAS-Analyzer SST_enhancer -o KAS-seq_SST_enhancers -s mm10 -e H3K27ac_enhancers.bed -p KAS-seq_peaks.bed -k KAS-seq.rep1.bam,KAS-seq.rep2.bam &
-o [prefix]: please input the prefix (basename) of 'KAS-Analyzer SST_enhancer' output files. DEFAULT: basename of enhancer file.
-t [threads]: please specify the number of threads used for single-stranded transcribing enhancers (SST enhancers) identification. DEFAULT: 1.
-s [assembly id]: please specify the genome assembly id. e.g. Human: hg18, hg19, hg38; Mouse: mm9, mm10, mm39; C.elegans: ce10, ce11; D.melanogaster: dm3, dm6; Rat: rn6, rn7; Zebra fish: danRer10, danRer11. REQUIRED.
-e [enhancer]: please specify the enhancer file used for single-stranded transcribing enhancers (SST enhancers) identification. Enhancer file can be H3K27ac, P300 or Med1 ChIP-seq peaks file. REQUIRED.
-p [peaks]: please specify the (sp)KAS-seq peaks file. REQUIRED.
-k [KAS-seq]: please specify the indexed bam file of KAS-seq data used for single-stranded transcribing enhancers (SST enhancers) identification. e.g. KAS-seq.rep1.bam,KAS-seq.rep2.bam. REQUIRED.
-h/--help: print this help and exit.
Note: The 'KAS-Analyzer ss_enhancer' shell script is applied to identify single-stranded transcribing enhancers (SST enhancers).
motif
Identify enriched TF binding motifs on ss_enhancers.
Usage: KAS-Analyzer motif [ -h/--help ] [ -t threads ] [ -o prefix ] [ -s assembly id ] [ -e enhancer position file ] [ -c control position file ]
Example: nohup KAS-Analyzer motif -o KAS-seq_enhancers_motifs -t 10 -s mm10 -e enhancers.bed -c control_background_peaks.bed &
-t [threads]: please specify the number of threads used for enriched TF motifs on transcribing enhancers. DEFAULT: 1.
-s [assembly id]: please specify the genome assembly id of enhancers regulatory elements. -s [assembly id]. e.g. Human: hg18, hg19, hg38; Mouse: mm9, mm10, mm39; C.elegans: ce10, ce11; D.melanogaster: dm3, dm6; Rat: rn6, rn7; Zebra fish: danRer10, danRer11. REQUIRED.
-o [prefix]: please input the prefix (basename) of 'KAS-Analyzer motif' output files. Default: basename of enhancers file.
-e [enhaner]: please specify the enhancers position file in bed format. REQUIRED.
-c [control]: please specify the control position file in bed format, which was used as background for enriched TF motifs idenfication on enhancers. OPTIONAL
-h/--help: print this help and exit.
Note: The 'KAS-Analyzer motif' shell script is applied to identify enriched TF motifs on transcribing enhancers identified by KAS-seq data. For more detials, please refer to http://homer.ucsd.edu/homer/ngs/peakMotifs.html
Termination length sub-commands:
termilength
Calculate the transcription termination length of protein coding genes.
Usage: KAS-Analyzer termilength [ -h/--help ] [ -o prefix ] [ -t threads ] [ -b bin size ] [ -g assembly id ] [ -p peaks ] [ -l labels ] [ -k KAS-seq ]
Example: nohup KAS-Analyzer termilength -o KAS-seq_termination_length -t 10 -g mm10 -p peaks.txt -l labels.txt -k KAS-seq.txt &
-o [prefix]: please input the prefix (basename) of 'KAS-Analyzer termilength' output files. REQUIET.
-t [threads]: please specify the number of threads. DEFAULT: 1.
-b [bin size]: please specify the size of sliding bins used for transcription termination length calculation. DEFAULT:100.
-p [peaks]: please input the text file containing the peaks file of (sp)KAS-seq data. REQUIET.
Example:
WT.peaks.rep1.bed
WT.peaks.rep2.bed ---peaks.txt
-s [assembly id]: please specify the genome assembly id of (sp)KAS-seq data. -g [assembly id]. e.g. Human: hg18, hg19, hg38; Mouse: mm9, mm10, mm39; C.elegans: ce10, ce11; D.melanogaster: dm3, dm6; Rat: rn6, rn7; Zebra fish: danRer10, danRer11. REQUIRED.
-l [labels]: please input the text file containing the labels of (sp)KAS-seq data that show in 'KAS-Analyzer termination_length' output files. DEFAULT: basename of KAS-seq file.
Example:
WT.rep1
WT.rep2 ---labels.txt
-k [KAS-seq]: please input the text file containing indexed (sp)KAS-seq bam files used for transcription termination length calculation. OPTIONAL.
Example:
KAS-seq.rep1.bam
KAS-seq.rep2.bam ---KAS-seq.txt
-h/--help: print this help and exit.
Note: The 'KAS-Analyzer termilength' shell script is applied to calculate the transcription termination length of protein coding genes.
KAS-seq index sub-commands:
index
Calculate the pausing or termination index.
Usage: KAS-Analyzer index [ -h/--help ] [ -o prefix ] [ -t threads ] [ -s assembly id ] [ -i index types ] [ -l labels ] [ -k KAS-seq ]
Example: nohup KAS-Analyzer index -o KAS-seq_pausing_index -t 10 -s hg19 -i pausing -l labels.txt -k KAS-seq.txt &
-o [prefix]: please input the prefix (basename) of 'KAS-Analyzer index' output files. REQUIRED.
-t [threads]: please specify the number of threads used for the pausing or termination index calculation. DEFAULT: 1.
-s [assembly id]: please specify the genome assembly id of KAS-seq data. -s [assembly id]. e.g. Human: hg18, hg19, hg38; Mouse: mm9, mm10, mm39; C.elegans: ce10, ce11; D.melanogaster: dm3, dm6; Rat: rn6, rn7; Zebra fish: danRer10, danRer11. REQUIRED.
-i [index]: please specify the index type you want to calculate. e.g. pausing or termination. REQUIRED.
-l [labels]: please input the text file containing the labels of (sp)KAS-seq that show in 'KAS-Analyzer index' output files. DEFAULT: basename of KAS-seq files.
Example:
WT_rep1
WT.rep2 ---labels.txt
-k [KAS-seq]: please input the text file containing indexed (sp)KAS-seq bam files used for pausing or termination index calculation. REQUIRED.
Example:
KAS-seq_WT_rep1.bam
KAS-seq_WT_rep2.bam ---KAS-seq.txt
-h/--help: print this help and exit.
Note: The 'KAS-Analyzer index' shell script is applied to calculate the pausing or termination index.
KASindex
Calculate KAS-seq index on promoter, genebody, genes or custom regions.
Usage: KAS-Analyzer KASindex [ -h/--help ] [ -t threads ] [ -o prefix ] [ -s assembly id ] [ -r regions ] [ -p peaks ] [ -l labels ] [ -k KAS-seq ]
Example: nohup KAS-Analyzer KASindex -o KAS-seq_index -t 10 -s mm10 -r TSS -l labels.txt -k KAS-seq_data.txt &
-t [threads]: please specify the number of threads used for calculating KAS expression. DEFAULT: 1.
-o [prefix]: please input the prefix (basename) of 'KAS-Analyzer KASexpre' output files. Default: basename of KAS-seq data.
-s [assembly id]: please specify the genome assembly id of KAS-seq data. -s [assembly id]. e.g. Human: hg18, hg19, hg38; Mouse: mm9, mm10, mm39; C.elegans: ce10, ce11; D.melanogaster: dm3, dm6; Rat: rn6, rn7; Zebra fish: danRer10, danRer11. REQUIRED.
-r [regions]: please specify the types of genomic regions. e.g. promoter, genebody, gene or peak. REQUIRED.
-p [peaks]: please specify the custom regions file used for KAS index calculation. OPTIONAL.
-l [labels]: please input the text file containing the labels of (sp)KAS-seq data that show in output files. REQUIRED.
Example:
WT.rep1
WT.rep2
KO.rep1
KO.rep2 ---labels.txt
-k [KAS-seq]: please input the text file containing the indexed bam files of (sp)KAS-seq data that used to calculate KAS expression. REQUIRED.
Example:
KAS-seq_WT.rep1.bam
KAS-seq_WT.rep2.bam
KAS-seq_KO.rep1.bam
KAS-seq_KO.rep2.bam ---KAS-seq_data.txt
-h/--help: print this help and exit.
Note: The 'KAS-Analyzer KASexpre' shell script is applied to calculate KAS-seq index on promoter, genebody, genes or custom regions.
General help sub-commands:
–help
Print this help menu.
–version
Print the version of KAS-Analyzer you are using.
–contact
Feature requests, bugs, mailing lists, etc.